内殻準位の絶対束縛エネルギーの計算方法を計算事例を紹介しながら、以下に説明します。
まずC![]() H
H![]() 分子の炭素原子の1s状態の束縛エネルギーの計算を実行します。
分子の炭素原子の1s状態の束縛エネルギーの計算を実行します。
始状態計算は以下のように実行されます。
% mpirun -np 4 ./openmx C2H2.datここで入力ファイル「C2H2.dat」はディレクトリ「work」中に収容されています。 始状態計算では以下のキーワードを指定する必要があります。
scf.coulomb.cutoff on # default = off
scf.SpinPolarization on or nc # default = off
「scf.coulomb.cutoff=on」の場合、
厳密クーロン遮蔽法 [91]を用いてa, b, c軸の三つの方向のすべてに沿って
スーパーセル間の古典的なクーロン相互作用がカットオフされます。
始状態と終状態の計算のどちらの計算においても、
キーワード「scf.SpinPolarization」に「on」または「nc」を指定する必要があります。
終状態計算では内殻ホールの生成の後に系はスピン分極するため、
始状態と終状態の計算の両方で同一のオプションを使用して下さい。
内殻準位の絶対束縛エネルギーを計算するため内殻状態を含む擬ポテンシャルを使用して下さい。
入力ファイル「C2H2.dat」では以下の擬ポテンシャルが指定されます。
<Definition.of.Atomic.Species
H H7.0-s3p2 H_PBE19
C C7.0_1s-s4p3d2 C_PBE19_1s
C1 C7.0_1s_CH-s4p3d2 C_PBE19_1s
Definition.of.Atomic.Species>
擬ポテンシャルC_PBE19_1s.vpsは1s, 2s, 2p状態を価電子状態として含みます。
C7.0_1s.pao と C7.0_1s_CH の基底セットをそれぞれ始状態と終状態の計算に用います。
終状態の計算では内殻ホールによって動径波動関数が内殻ホールが無い状態に比べて大幅に変形しています。
収束した計算結果を得るためには内殻ホールが存在した状態に対して最適化された基底セットを用いる必要があります。
終状態計算の擬ポテンシャルと基底セットは以下のウェブサイトで利用可能です。
https://t-ozaki.issp.u-tokyo.ac.jp/vps_pao_core2019/
本ウェブサイト上では現在七つの元素 B, C, N, O, Si, S, Ge, Pt のみのデータが利用可能です。
近い将来に他の元素の擬ポテンシャルと基底関数セットも公開する予定です。
始状態の計算では幾何学構造は以下のように指定されています。
Atoms.Number 4
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.6005 0.000 0.000 3.0 3.0
2 C -0.6005 0.000 0.000 3.0 3.0
3 H 1.8015 0.000 0.000 0.5 0.5
4 H -1.8015 0.000 0.000 0.5 0.5
Atoms.SpeciesAndCoordinates>
原子1の元素種は「C」で、この原子に対して始状態ではC7.0_1s.paoを基底セットに割り当てます。
次に説明する終状態の計算では、原子1に対して1s状態の内殻ホールを生成することになります。
終状態 計算は以下のように実行します。
% mpirun -np 4 ./openmx C2H2-CH.dat入力ファイル「C2H2-CH.dat」はディレクトリ「work」中に収容されています。 終状態の計算では幾何構造は以下のように指定されています。
Atoms.Number 4
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C1 0.6005 0.000 0.000 3.0 3.0
2 C -0.6005 0.000 0.000 3.0 3.0
3 H 1.8015 0.000 0.000 0.5 0.5
4 H -1.8015 0.000 0.000 0.5 0.5
Atoms.SpeciesAndCoordinates>
原子位置は始状態計算と厳密に同一です。これは励起過程の際での原子緩和は考慮されないことを意味します。
原子1の元素種は「C1」で、この原子に対して終状態では基底関数セットとして「C7.0_1s_CH.pao」を割り当てていることに
注意して下さい。
終状態計算では以下のキーワードを指定する必要があります。
scf.system.charge 1.0 # default=0.0
scf.coulomb.cutoff on # default = off
scf.core.hole on # default = off
<core.hole.state
1 s 1
core.hole.state>
分子系の終状態が![]() 電子系であることを考慮し、キーワード「scf.system.charge」で電子数を1つ減らします。
次に、スーパーセル間の内殻ホール間の偽の相互作用を回避するため、
キーワード「scf.coulomb.cutoff」を用いて厳密クーロンカットオフ法[91]を適用します。
もし、厳密クーロンカットオフ法が適用されない場合には
周期的な帯電系でのクーロン発散を避けるため、電荷補償のために反対電荷を持つ一様な電荷が自動的に導入されます。
この扱いでは偽の相互作用が生じるため、始状態と終状態の間で全エネルギーの直接の比較ができなくなります。
電子系であることを考慮し、キーワード「scf.system.charge」で電子数を1つ減らします。
次に、スーパーセル間の内殻ホール間の偽の相互作用を回避するため、
キーワード「scf.coulomb.cutoff」を用いて厳密クーロンカットオフ法[91]を適用します。
もし、厳密クーロンカットオフ法が適用されない場合には
周期的な帯電系でのクーロン発散を避けるため、電荷補償のために反対電荷を持つ一様な電荷が自動的に導入されます。
この扱いでは偽の相互作用が生じるため、始状態と終状態の間で全エネルギーの直接の比較ができなくなります。
| コリニアの場合 | ||||||||
|
|
1: |
2: |
||||||
|
|
1: |
2: |
3: |
4: |
5: |
6: |
||
|
|
1:
|
2:
|
3:
|
4:
|
5:
|
|||
|
6:
|
7:
|
8:
|
9:
|
10:
|
||||
|
|
1:
|
2:
|
3:
|
4:
|
5:
|
6:
|
7:
|
|
|
8:
|
9:
|
10:
|
11:
|
12:
|
13:
|
14:
|
||
| ノンコリニアの場合 | ||||||||
|
|
1: |
2: |
||||||
|
|
1: |
2: |
3: |
4: |
5: |
6: |
||
|
|
1: |
2: |
3: |
4: |
5: |
6: |
||
|
7: |
8: |
9: |
10: |
|||||
|
|
1: |
2: |
3: |
4: |
5: |
6: |
7: |
8: |
|
9: |
10: |
11: |
12: |
13: |
14: |
キーワード「core.hole.state」により内殻ホールの生成を行う内殻状態を指定します。
最初の数はキーワード「Atoms.SpeciesAndCoordinates」に指定される原子の通し番号です。
二番目の記号は対象の![]() チャネルを指定し、「
チャネルを指定し、「![]() 」、「
」、「![]() 」、「
」、「![]() 」、「
」、「![]() 」を指定できます。
最後の数字は軌道インデックスを指定し、1から
」を指定できます。
最後の数字は軌道インデックスを指定し、1から![]() までを指定できます。
軌道インデックスと内殻状態の関係は表 12に与えられています。
内殻ホールの生成を行う対象となる内殻状態は価電子状態として擬ポテンシャルに含まれた
までを指定できます。
軌道インデックスと内殻状態の関係は表 12に与えられています。
内殻ホールの生成を行う対象となる内殻状態は価電子状態として擬ポテンシャルに含まれた
![]() チャネルの最下位の状態であることに注意してください。
C
チャネルの最下位の状態であることに注意してください。
C![]() H
H![]() 分子の場合は、擬ポテンシャルC_PBE19_1s.vpsは1s状態を価電状態として含みます。
よって以下の指定は原子1の
分子の場合は、擬ポテンシャルC_PBE19_1s.vpsは1s状態を価電状態として含みます。
よって以下の指定は原子1の
![]() の状態に対して内殻ホールの生成を行う指定となります。
の状態に対して内殻ホールの生成を行う指定となります。
<core.hole.state
1 s 1
core.hole.state>
始状態と終状態の計算が終わると、出力ファイルから以下の全エネルギーが得られます。
Initial state: -76.787732114928 (Hartree)
Final state: -66.084858926233 (Hartree)
そして、式 (12)を用いて、束縛エネルギーは以下のとおり計算できます。
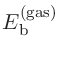 |
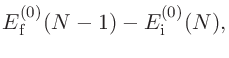 |
||