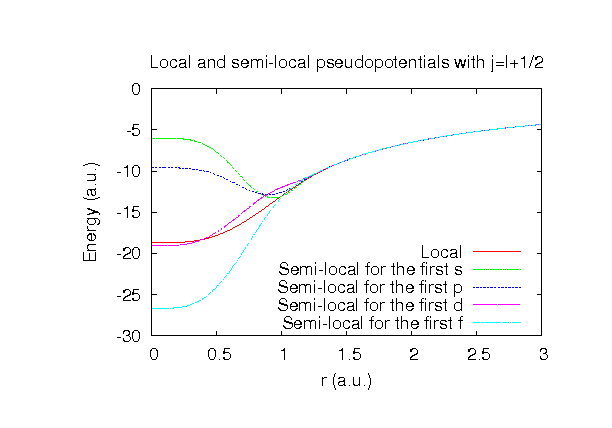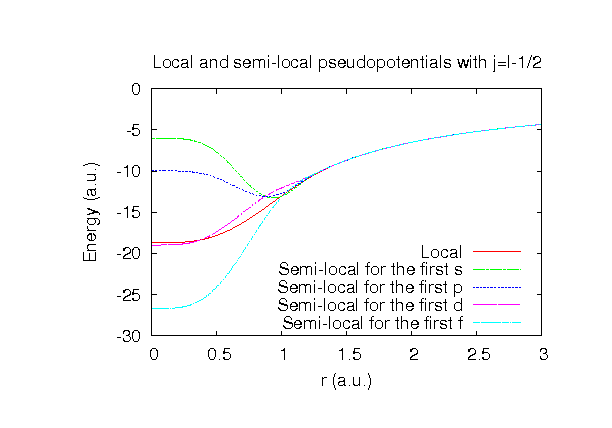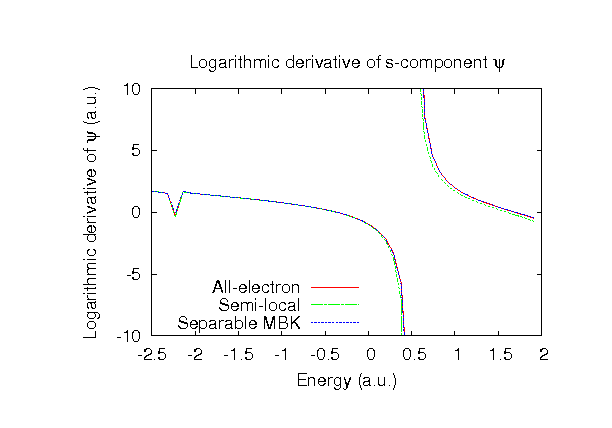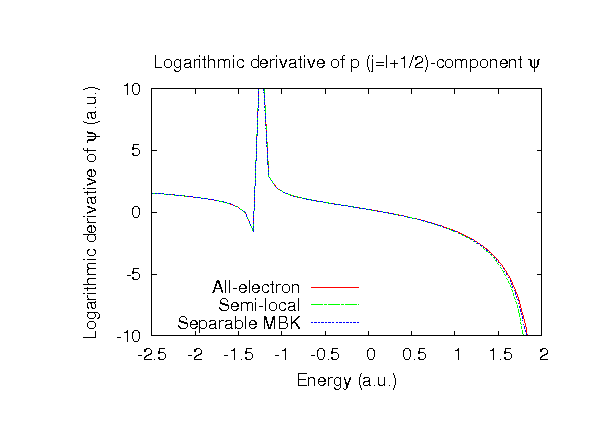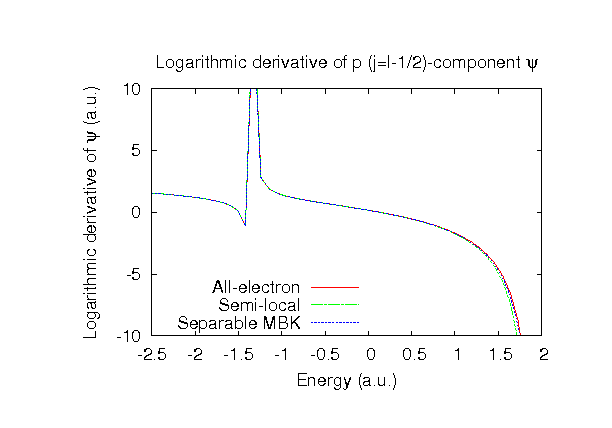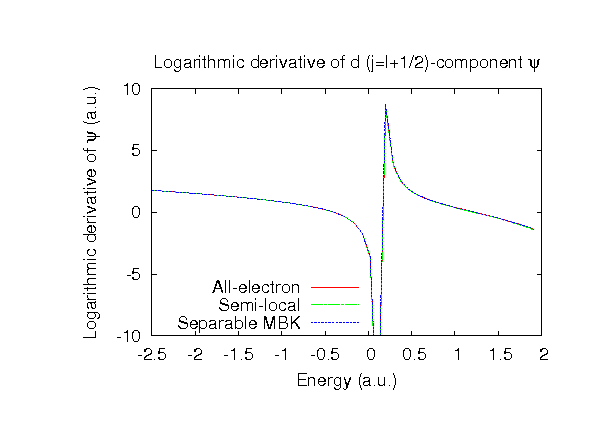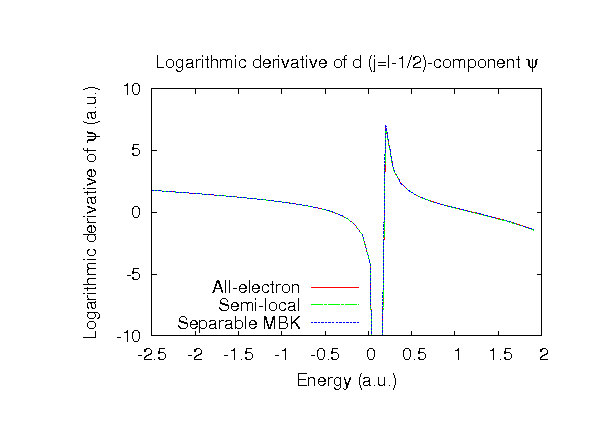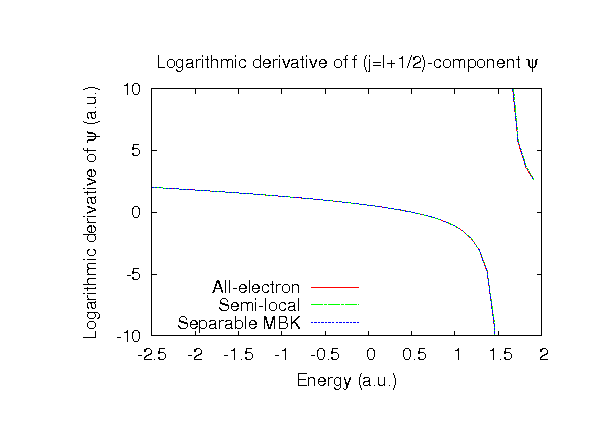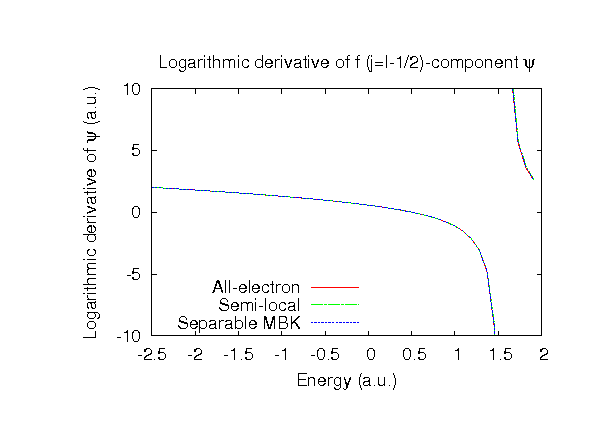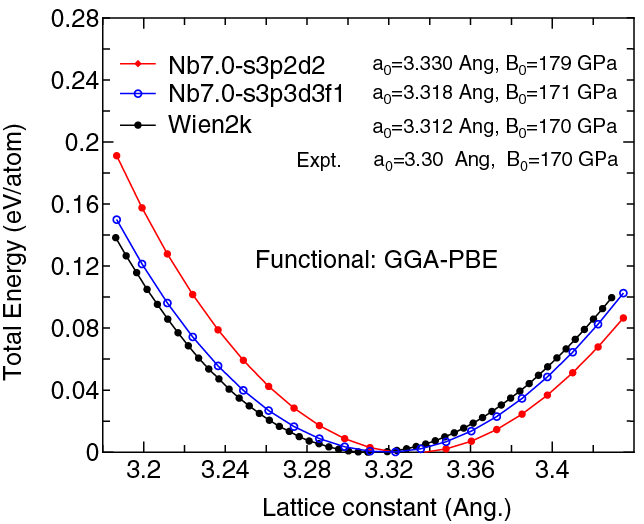
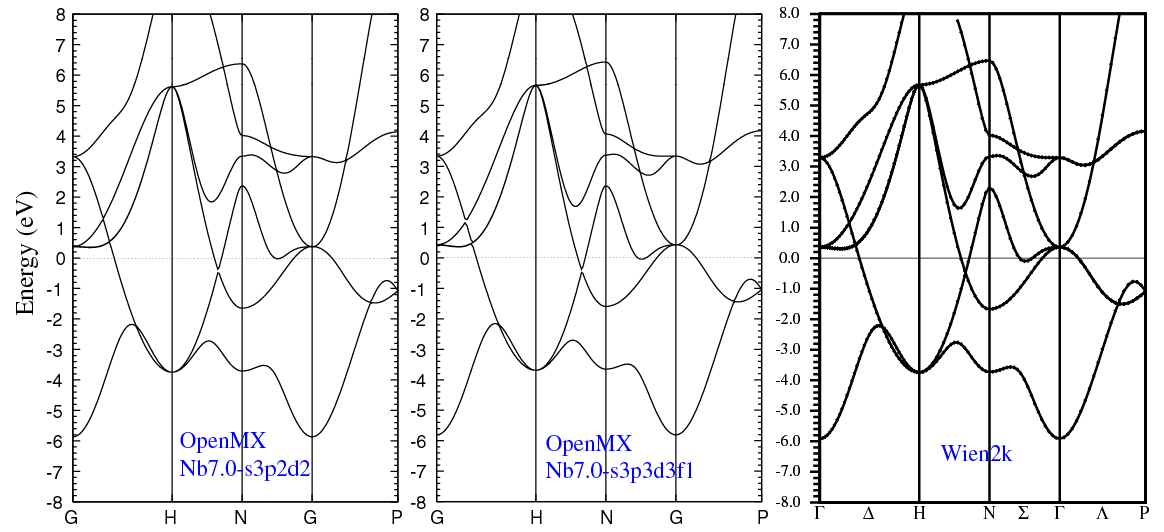
| |
|
|
|
| Equilibrium bond length (Ang.) | Atomization energy (eV) | Atomization energy (couterpoise corrected) (eV) | |
| |
|
|
|
| Nb9.0-s3p2d2 | 2.164 | 4.16 | 4.15 |
| Nb9.0-s3p3d3f1 | 2.120 | 4.48 | 4.45 |
| Other calc. | 2.099a | 3.912 a | - |
| Expt. | 2.08 b | 5.48 b | - |
| |
|
|
|