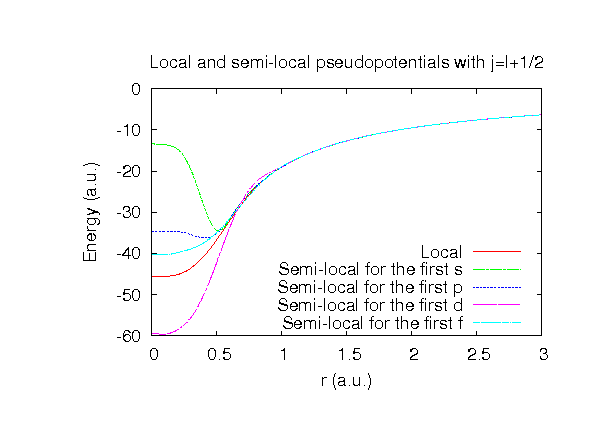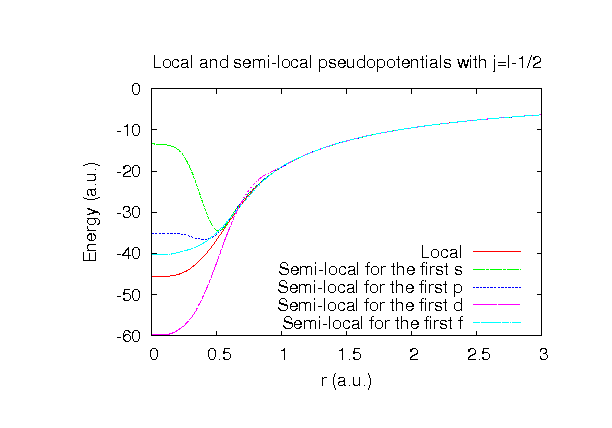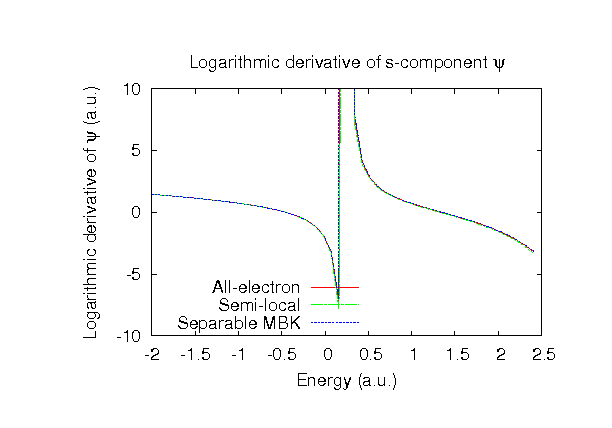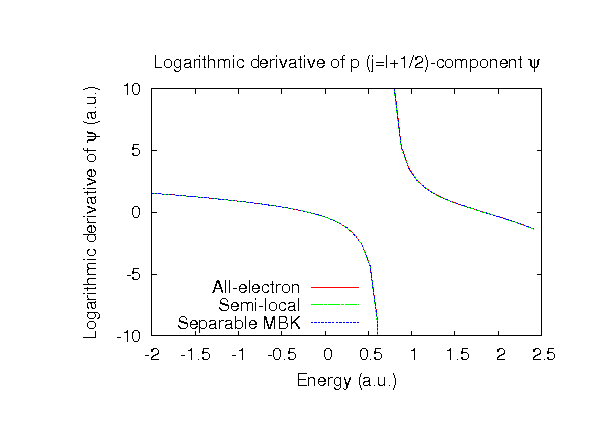
| |
|
|
|
| Equilibrium bond length (Ang.) | Atomization energy (eV) | Atomization energy (couterpoise corrected) (eV) | |
| |
|
|
|
| Cu10.0H-s3p3d3 | 2.247 | 2.156 | 2.152 |
| Cu10.0H-s3p3d3f1 | 2.241 | 2.183 | 2.180 |
| Other calc. | 2.23 a | 2.28 a | - |
| Expt. | 2.2197 b | 1.83 b | - |
| |
|
|
|