The calculation of Energy vs. lattice constant is supported by the following keywords:
MD.Type EvsLC #
MD.EvsLC.Step 0.4 # default=0.4%
MD.maxIter 32 # default=1
MD.EvsLC.flag 1 1 1 # default=1 1 1
# (0: fixed, 1:expansion, -1:contraction)
When 'MD.Type' is set to 'EvsLC', the total energy is calculated step by step by
changing unit cell vectors, a, b, and c.
The change of unit cell vectors is done uniformly by
expanding them by a percentage, where the reference is the initial vectors,
specified with 'MD.EvsLC.Step'. The number of steps is specified by the keyword
'MD.maxIter'. If you want to fix some of lattice vectors, the keyword 'MD.EvsLC.flag'
is available, where the default setting is '1 1 1' corresponding to the uniform expansion
of a-, b-, c-axes, respectively. The flag '0' means no change of the corresponding axis,
and '-1' the uniform contraction.
After the calculation, you will obtain a file 'System.Name.EvsLC', where
'System.Name' is 'System.Name'. The columns in the file 'System.Name.EvsLC' are arranged in order
of 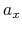 ,
, 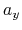 ,
, 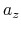 ,
, 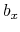 ,
, 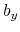 ,
, 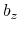 ,
, 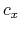 ,
, 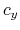 ,
, 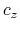 in Å,
and the total energy in Hartree, where
in Å,
and the total energy in Hartree, where 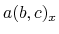 ,
, 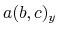 , and
, and 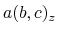 are
are 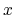 -,
-, 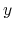 -, and
-, and 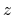 -coordinates of the a(b,c) vector, respectively.
As an example, calculation of Energy vs. lattice for the fcc Mn bulk
is shown in Fig. 88, where the equilibrium lattice constant and
bulk modulus were evaluated by fitting the data to the Murnaghan equation of state
with a code 'murn.f' provided on the website [148].
-coordinates of the a(b,c) vector, respectively.
As an example, calculation of Energy vs. lattice for the fcc Mn bulk
is shown in Fig. 88, where the equilibrium lattice constant and
bulk modulus were evaluated by fitting the data to the Murnaghan equation of state
with a code 'murn.f' provided on the website [148].
Figure 88:
Total energy vs. lattice constant for the fcc Mn bulk calculated by the
keyword 'EvsLC'. The input file used for the calculation is
'Mnfcc-EvsLC.dat' in the directory 'work'.
|
|
![\includegraphics[width=13.0cm]{EvsLC.eps}](img608.png)