ダイヤモンド構造の炭素を例として、DOSの計算方法を説明します。 「work」ディレクトリ内のファイル「Cdia.dat」には、次の様にDOS計算のためのキーワードが指定されています。
Dos.fileout on
Dos.Erange -25.0 20.0
Dos.Kgrid 12 12 12
キーワード「Dos.Erange」で指定された最初と2番目の数値は、それぞれDOS計算のためのエネルギー範囲(eV)の下限と上限であり、
エネルギーの原点(0.0)は化学ポテンシャルに対応します。
またキーワード「Dos.Kgrid」で指定された数値(n1,n2,n3)は、DOS計算での第1ブリルアンゾーンを離散化するための
グリッド数です。
次にOpenMXを以下の様に、通常実行します。
% ./openmx Cdia.dat
ここでは単一コアを使用した計算例を示しましたが、もちろん並列計算も実行可能です。
計算が正常に終了すると、「work」ディレクトリに「cdia.Dos.val」および「cdia.Dos.vec」の2つのファイルが生成されます。
「cdia.Dos.val」にはテキスト形式で固有値が、「cdia.Dos.vec」にはバイナリ形式で固有ベクトルが保存されています。
このDOS計算は、O(DOS計算用のプログラムパッケージをコンパイルして下さい。「source」ディレクトリ内で、次のコマンドでコンパイルします。
% make DosMain
コンパイルが正常に終了すると、「source」ディレクトリ内に実行ファイル「DosMain」が生成されます。
「DosMain」を「work」ディレクトリにコピーして、「work」ディレクトリに移動してください。
このプログラム「DosMain」を使って先の2個のファイル「cdia.Dos.val」および「cdia.Dos.vec」から
全状態密度(DOS)と射影したDOS(PDOS)を次の様に計算します。
% ./DosMain cdia.Dos.val cdia.Dos.vec
その際に、プログラムから対話形式で次のように質問されることでしょう。
% ./DosMain cdia.Dos.val cdia.Dos.vec
Max of Spe_Total_CNO = 8
1 1 101 102 103 101 102 103
<cdia.Dos.val>
<cdia>
Which method do you use?, Tetrahedron(1), Gaussian Broadeninig(2)
1
Do you want Dos(1) or PDos(2)?
2
Number of atoms=2
Which atoms for PDOS : (1,...,2), ex 1 2
1
pdos_n=1
1
<Spectra_Tetrahedron> start
Spe_Num_Relation 0 0 1
Spe_Num_Relation 0 1 1
Spe_Num_Relation 0 2 101
Spe_Num_Relation 0 3 102
Spe_Num_Relation 0 4 103
Spe_Num_Relation 0 5 101
Spe_Num_Relation 0 6 102
Spe_Num_Relation 0 7 103
make cdia.PDOS.Tetrahedron.atom1.s1
make cdia.PDOS.Tetrahedron.atom1.p1
make cdia.PDOS.Tetrahedron.atom1.p2
make cdia.PDOS.Tetrahedron.atom1.p3
make cdia.PDOS.Tetrahedron.atom1
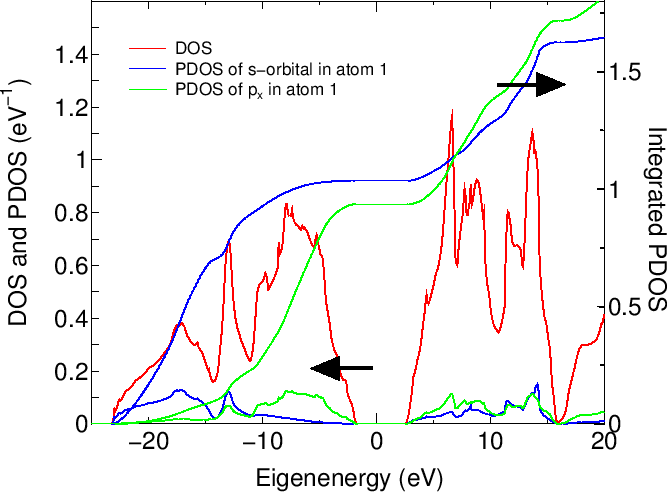
DOSの計算には四面体法(tetrahedron method)[48]もしくはガウシアンブロードニング法(Gaussian broadening method)が選択できます。
またユーザーはDOSまたはPDOSを選ぶことができます。PDOSの計算を選択する場合は、PDOSを評価する原子を選択してください。
この場合には、選択した原子の軌道(s, px(p1), py(p2), pz(p3),..)上に射影したPDOSが各ファイルに出力されます。
これらのファイルでは、最初と二番目の列は、エネルギー(eV)、およびDOS(eV
 |