After finishing your first calculation or achieving the self consistency, you may want to continue the calculation or to calculate density of states, band dispersion, molecular orbitals, and etc. using the self consistent charge in order to save the computational time. To do this, a keyword 'scf.restart' is available.
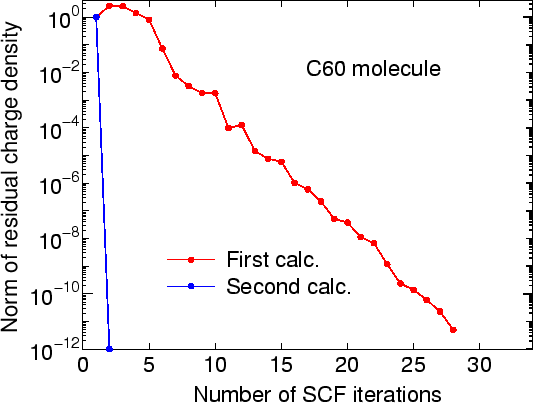
scf.restart on # on|off,default=offWhen the keyword 'scf.restart' is switched on, restart files generated by your first calculation will be used as the input Hamiltonian or charge density in the second calculation, while 'System.Name' in the second calculation should be the same as in the first calculation. The restart files are stored in a directory '*_rst' below the 'work' directory, where * means System.Name. The restart files in the '*_rst' contain all the information for both the density matrix mixing schemes and k-space mixing schemes. So, it is also possible to use another mixing scheme in the second calculation. As an example, we illustrate the restarting procedure using an input file C60.dat which can be found in the directory 'work'. In Fig. 7, we see that the second calculation is accelerated due to the use of the restart file.
 |