



Next: 出力ファイル
Up: 入力ファイル
Previous: 例:メタン分子
Contents
Index
各キーワードについて下記に説明していきます。この章ではOpenMXのすべてのキーワードは網羅しておらず、また各キーワードについては対応する章で更に詳しく説明しています。本マニュアルの最後に記載されているインデックスをご参照頂ければ、各キーワードに関連したページ番号が分かります。
ファイル名
System.CurrrentDir
このキーワードにより、出力ファイルの出力ディレクトを指定します。デフォルト値は「./」です。
System.Name
このキーワードで出力ファイルのファイル名を指定します。
DATA.PATH
VPSおよびPAOディレクトリへのパスを下記のように指定します。
DATA.PATH ../DFT_DATA19 # default=../DFT_DATA19
絶対パス、相対パスとも指定可能です。デフォルト値は「../DFT_DATA19」です。
level.of.stdout
このキーワードにより、標準出力への出力情報のレベルを制御します。
「level.of.stdout=0」を指定した場合、最小限の情報が出力されます。
「level.of.stdout=1」の場合、最小限の出力に加えて追加の情報が出力されます。
「level.of.stdout=2」は開発者向けのオプションです。デフォルト値は1です。
level.of.fileout
このキーワードにより、出力ファイルへの出力情報のレベルを制御します。「level.of.fileout=0」を指定した場合、最小限の情報が出力されます(Gaussian cubeファイルおよびグリッドファイルの出力無し)。
「level.of.fileout=1」は標準的な出力レベルです。「level.of.fileout=2」の場合、標準の出力に加えて追加の情報が出力されます。デフォルト値は1です。
原子種の定義
Species.Number
このキーワードにより、系に含まれる原子種の数を指定します。
Definition.of.Atomic.Species
擬原子基底関数と擬ポテンシャルの両方のファイル名を明記することにより、原子種を指定して下さい。
これらのファイルはそれぞれ「DFT_DATA19/PAO」および「DFT_DATA19/VPS」ディレクトリに置かなければなりません。例えば以下のように指定します。
<Definition.of.Atomic.Species
H H5.0-s1>1p1>1 H_CA19
C C5.0-s1>1p1>1 C_CA19
Definition.of.Atomic.Species>
原子種の指定は「 Definition.of.Atomic.Species」で始め、「Definition.of.Atomic.Species
Definition.of.Atomic.Species」で始め、「Definition.of.Atomic.Species 」で終わらなければなりません。1番目の列では任意の原子種の名前を指定します。
この名前は、後述する
」で終わらなければなりません。1番目の列では任意の原子種の名前を指定します。
この名前は、後述する
「Atoms.SpeciesAndCoordinates」キーワードにて原子座標を指定するのに使います。
2番目の列では擬原子基底関数の拡張子無しのファイル名と、プリミティブ基底関数の数および
縮約された基底関数の数を指定します。
例では基底関数は「H4.0-s11p11」となっていますが、H4.0が「DFT_DATA19/PAO」ディレクトリにある
擬原子基底関数の拡張子無しのファイル名を示し、s11は、1つのs軌道のプリミティブ軌道から1つの
最適化された軌道が作られていることに対応します。つまり縮約は行わないことを意味します。
縮約が無い場合、「s11」と記述する代わりに「s1」という簡単な記法を使用できます。
つまり、「H4.0-s1p1」は「H4.0-s11p11」と同等ということです。
3番目の列では擬ポテンシャルの拡張無しのファイル名を指定します。
このファイルは「DFT_DATA19/VPS」ディレクトリ内に存在しなければなりません。
同一元素に対して異なる基底関数と擬ポテンシャルを割り当てることで、異なる原子種を定義することもできます。
例えば複数の原子種を以下のように指定できます。
<Definition.of.Atomic.Species
H1 H5.0-s1p1 H_CA19
H2 H5.0-s2p2d1 H_CA19
C1 C5.0-s2p2 C_CA19
C2 C5.0-s2p2d2 C_CA19
Definition.of.Atomic.Species>
この柔軟性の高い記述法を活用し、系の特性を決定している領域に属する原子には高精度の基底関数を割り当て、
不活性な領域に属する原子には低精度の基底関数を割り当てることも可能であり、
計算量の削減には有効な手段です。
原子
Atoms.Number
系の合計原子数を「Atoms.Number」キーワードで指定します。
Atoms.SpeciesAndCoordinates.Unit
原子座標の単位を「Atoms.SpeciesAndCoordinates.Unit」キーワードで指定します。
オングストロームの場合は「Ang」、原子単位の場合は「AU」を指定します。「FRAC」と記述することで、規格化座標を指定することも可能です。
規格化座標の場合には、後述する「Atoms.UnitVectors」のキーワードで与えられた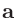 、
、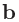 、
、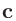 ベクトルを基底として、
座標を指定します。その際には0.0〜1.0の範囲で座標が設定可能で、この範囲を超える座標は入力ファイルの読み込み後に自動的に調整されます。
ベクトルを基底として、
座標を指定します。その際には0.0〜1.0の範囲で座標が設定可能で、この範囲を超える座標は入力ファイルの読み込み後に自動的に調整されます。
Atoms.SpeciesAndCoordinates
原子座標および各スピン毎の電子数を「Atoms.SpeciesAndCoordinates」キーワードで以下の例のように指定します。
<Atoms.SpeciesAndCoordinates
1 C 0.000000 0.000000 0.000000 2.0 2.0
2 H -0.889981 -0.629312 0.000000 0.5 0.5
3 H 0.000000 0.629312 -0.889981 0.5 0.5
4 H 0.000000 0.629312 0.889981 0.5 0.5
5 H 0.889981 -0.629312 0.000000 0.5 0.5
Atoms.SpeciesAndCoordinates>
記述は「Atoms.SpeciesAndCoordinates」で始まり、「Atoms.SpeciesAndCoordinates」で終わります。
1列目には原子を特定する連番を記述します。
2列目には事前に「Definition.of.Atomic.Species」の1列目で定義した原子種を指定します。
3〜5列目には、それぞれ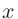 、
、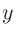 、
、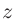 座標を指定します。
座標を指定します。
「Atoms.SpeciesAndCoordinates.Unit」キーワードで「FRAC」を指定している場合は3〜5列目はそれぞれ、、軸の
規格化座標になり、値は0.0〜1.0の範囲で指定します。
この範囲を外れる数値は入力ファイルが読み込まれる段階で自動的に調整されます。
6および7列目には、各原子のアップスピンとダウンスピン状態のそれぞれの初期電子数を設定します。
アップスピン電子数とダウンスピン電子数の合計は原子の価電子の数に等しくなるようにします。
原子の価電子数は表1と表2に記載されています.
「LSDA-CA」、「LSDA-PW」、あるいは「GGA-PBE」を使用してスピン分極系を計算する場合、6および7列目の指定によって
各原子に初期スピンを指定することができます。これにより強磁性状態や反強磁性状態などの計算が可能です。
またSCFの収束を加速するためには、この初期スピン配置は基底状態に近い配置からスタートすることが重要です。
Atoms.UnitVectors.Unit
単位胞(ユニットセル)におけるベクトルの単位を「Atoms.UnitVectors.Unit」キーワードで指定します。
オングストロームの場合は「Ang」、原子単位の場合は「AU」を指定します。
Atoms.UnitVectors
単位胞のベクトル、、をAtoms.UnitVectorsキーワードで以下の様に指定します。
<Atoms.UnitVectors
10.0 0.0 0.0
0.0 10.0 0.0
0.0 0.0 10.0
Atoms.UnitVectors>
記述は「Atoms.UnitVectors」で始まり、「Atoms.UnitVectors」で終わります。
第1〜3行はそれぞれ単位胞のベクトル、、に対応します。
クラスタ計算の際にこのキーワードが指定されないと周期的に配置された孤立系の間で基底関数が重ならないように、
単位胞の大きさが自動的に決定されます。
詳細に関しては「セルサイズの自動決定」の章もご参照下さい。
SCFまたは電子系
scf.XcType
交換相関ポテンシャルを「scf.XcType」キーワードで指定します。
OpenMX Ver. 3.9においては「LDA」、「LSDA-CA」、「LSDA-PW」、「GGA-PBE」から選択できます。
ここで「LSDA-CA」はCeperley-Alder [2]の局所スピン密度関数、「LSDA-PW」は 、
そのGGA形式において密度勾配をゼロとしたPerdew-Wang局所スピン密度関数 [4]です。
「GGA-PBE」はPerdewらが提案するGGA汎関数 [5]です。
scf.SpinPolarization
電子系の非スピン分極あるいはスピン分極は「scf.SpinPolarization」キーワードで指定します。
スピン分極の計算を行う場合は「ON」を指定し、非スピン分極の計算を行う場合は「OFF」を指定します。
「scf.XcType」キーワードで「LDA」を使用する場合は「scf.SpinPolarization」を「OFF」に設定して下さい。
前述の2つのオプションの他、ノンコリニアDFT計算を行う場合には「NC」というオプションを指定して下さい。
この計算については「ノンコリニアDFT」の章もご参照下さい。
scf.partialCoreCorrection
交換相関エネルギーおよびそのポテンシャルの計算において部分内殻補正(Partial Core Correction、PCC)を行う場合には、
「scf.partialCoreCorrection」キーワードを指定します。
これを「ON」に指定するとPCCを行い、「OFF」の場合にはPCCを行いません。
通常は、このフラグは「ON」にするべきです。
何故ならば、PCC擬ポテンシャルを使用する場合にはPCCを行うべきであり、また非PCC擬ポテンシャルの場合でも
PCC電荷がゼロなので結果には影響を及ぼさないからです。デフォルトは「ON」です。
scf.Hubbard.U
LDA+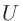 およびGGA+計算の場合、「scf.Hubbard.U」キーワードを「ON」に設定して下さい(ON
およびGGA+計算の場合、「scf.Hubbard.U」キーワードを「ON」に設定して下さい(ON OFF)。デフォルトでは「OFF」に設定されています。
OFF)。デフォルトでは「OFF」に設定されています。
scf.Hubbard.Occupation
LDA+法では、「scf.Hubbard.Occupation」キーワードを指定することで、「onsite」、「full」および「dual」の3つの占有数演算子から選択することができます。
Hubbard.U.values
下記のキーワードにより各原子種の軌道に対する実効的なUの値を定義します。
<Hubbard.U.values # eV
Ni 1s 0.0 2s 0.0 1p 0.0 2p 0.0 1d 4.0 2d 0.0
O 1s 0.0 2s 0.0 1p 0.0 2p 0.0 1d 0.0
Hubbard.U.values>
記述は「Hubbard.U.values」で始まり、「Hubbard.U.values」で終わります。
「Definition.of.Atomic.Species」で指定した全ての基底軌道に対して、上記の形式にて実効的なUの値を設定しなければなりません。
「1s」や「2s」はそれぞれ割り当てられた最初と二番目の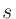 軌道を示し、「1s」の後にくる数字が最初の軌道に対する実効的なU値(eV)です。
同様の規則が、
軌道を示し、「1s」の後にくる数字が最初の軌道に対する実効的なU値(eV)です。
同様の規則が、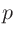 および
および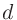 軌道にも当てはまります。
軌道にも当てはまります。
scf.Constraint.NC.Spin
スピン磁気モーメントのノンコリニア方位を制御する制約条件付DFT計算を行う場合には「scf.Constraint.NC.Spin」キーワードを「ON」に設定して下さい。
このキーワードは「ON」か「OFF」に設定することができます。
scf.Constraint.NC.Spin.v
スピン磁気モーメントのノンコリニア方位を制御する制約条件付DFT計算を行う場合には、
「scf.Constraint.NC.Spin.v」キーワードによって
ペナルティ関数の前因子(eV)を指定します。
scf.ElectronicTemperature
「scf.ElectronicTemperature」キーワードにて電子温度(K)を設定します。デフォルトでは300(K)に設定されています。
scf.energycutoff
「scf.energycutoff」キーワードで積分グリッド間隔を決定するカットオフエネルギーを指定します。
この積分グリッドは差電子クーロンポテンシャルと交換相関ポテンシャルに対する行列要素の計算
および高速フーリエ変換(FFT)を用いたPoisson方程式の
解法のために使用されます。デフォルトでは150(Ryd)に設定されています。
scf.Ngrid
「scf.Ngrid」キーワードによってa、b、c軸を離散化させる際のグリッド数を指定します。
通常は「scf.energycutoff」で指定した値を使って実空間の離散化を行いますが、「scf.Ngrid」でグリッド数を指定した場合には、
「scf.energycutoff」ではなくこちらが優先されます。
scf.maxIter
SCFの最大反復回数を「scf.maxIter」キーワードで設定します。
SCF反復ループは、収束条件が満たされない場合でもこのキーワードで指定した回数で終了します。デフォルト値は40です。
scf.EigenvalueSolver
固有値問題の計算手法を「scf.EigenvalueSolver」で指定します。O(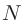 )分割統治法は「DC」、
局在自然軌道によるO()分割統治法は「DC-LNO」、O()クリロフ部分空間法は「Krylov」、
数値厳密な低次スケーリング法は「ON2」、クラスタ計算は「Cluster」、バンド計算は「Band」を指定します。
)分割統治法は「DC」、
局在自然軌道によるO()分割統治法は「DC-LNO」、O()クリロフ部分空間法は「Krylov」、
数値厳密な低次スケーリング法は「ON2」、クラスタ計算は「Cluster」、バンド計算は「Band」を指定します。
scf.Kgrid
OpenMXでは等間隔メッシュによりk空間の第1ブリルアンゾーンを離散します。
「scf.EigenvalueSolver」キーワードにより「Band」を指定した場合、k空間の第1ブリルアンゾーンを離散化するための
格子の数(n1, n2, n3)を「scf.Kgrid」キーワードで指定しなければなりません。
k空間の逆格子ベクトル
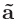 、
、
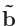 、
、
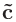 を離散化するために「n1 n2 n3」のように指定して下さい。
を離散化するために「n1 n2 n3」のように指定して下さい。
scf.ProExpn.VNA
中性原子ポテンシャルVNAを射影演算子で展開する場合 [42]には、「scf.ProExpn.VNA」キーワードを「ON」に設定し、
それ以外の場合は「OFF」を設定して下さい。デフォルトでは「ON」になっています。
scf.ProExpn.VNA ON # ON|OFF, default = ON
「scf.ProExpn.VNA=OFF」と設定されている場合、VNAポテンシャルの行列要素は実空間の離散グリッドを用いて計算されます。
scf.Mixing.Type
「scf.Mixing.Type」キーワードによって、SCF計算の次の反復ステップに入力される電子密度(もしくは密度行列)を生成するための電子密度混合法を指定します。
単純混合法(Simple)、
GR-Pulay法(Guaranteed-Reduction Pulay)[57]、
RMM-DIIS法 [58]、
Kerker法[59]、
RMM-DIISK法 [58]、
RMM-DIISV法 [58]
あるいは
RMM-DIISH法 [58]
のいずれかを指定することができます。
OpenMXの単純混合法は収束の速度を上げるために収束履歴を参照するように改良されています。
「GR-Pulay」、「RMM- DIIS」、「Kerker」、「RMM-DIISK」、「RMM-DIISV」あるいは「RMM-DIISH」を使用する際には、以下の点に注意することで
SCF計算の高速化が可能です。
- Pulay法に準拠する方法で混合を開始する前には、ある程度のSCF収束が達成されている必要があります。
そのため、やや大きめの「scf.Mixing.StartPulay」の値を使用します。
「scf.Mixing.StartPulay」の適切な値は10〜30です。
- 金属系の場合は「scf.ElectronicTemperature」で高温の電子温度を指定します。
「scf.ElectronicTemperature」があまりに低いと数値的に不安定な挙動がよく見られます。
- 「scf.Mixing.History」の値を大きくします。ほとんどの場合、「scf.Mixing.History=30」が妥当な値です。
前述した混合法のうち、多くの系に対して最もロバスト性が高い手法は「RMM-DIISK」です。
scf.Init.Mixing.Weight
単純混合法、GR-Pulay法, RMM-DIIS法、Kerker法、RMM-DIISK法、RMM-DIISV法、およびRMM-DIISH法で使用する初期の混合比を「scf.Init.Mixing.Weight」キーワードで指定します。
有効な範囲は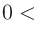 scf.Init.Mixing.Weight
scf.Init.Mixing.Weight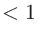 で、デフォルト値は0.3です。
で、デフォルト値は0.3です。
scf.Min.Mixing.Weight
単純混合法およびKerker混合法における混合比の下限を「scf.Min.Mixing.Weight」で指定します。デフォルトでは0.001に設定されています。
scf.Max.Mixing.Weight
単純混合法およびKerker混合法における混合比の上限を「scf.Max.Mixing.Weight」で指定します。デフォルトでは0.4に設定されています。
scf.Kerker.factor
KerkerおよびRMM-DIISK混合法におけるKerker因子を指定します。
このキーワードが指定されない場合、適切な値が自動的に設定されます。詳しくは「SCF収束」の章を参照して下さい。
scf.Mixing.History
GR-Pulay法[57]、 RMM-DIIS法[58]、RMM-DIISK法[58]、RMM-DIISV法[58]及びRMM-DIISH法[58]では、
SCFの次の反復ステップにおける入力電子密度(ハミルトニアン)を、過去のSCF反復の電子密度(ハミルトニアン)に基づき推定します。
「scf.Mixing.History」キーワードは、この推定に使用する過去のSCF反復ステップ数を指定します。
例えば、「scf.Mixing.History」を3に設定した場合、6回目のSCF反復では過去の第5、4、3ステップの電子密度(ハミルトニアン)が考慮されます。
30程度が適切な選択値です。
scf.Mixing.StartPulay
GR-Pulay法、RMM-DIIS法、Kerker法、RMM-DIISK法、RMM-DIISV法およびRMM-DIISH法を開始するSCFステップを
「scf.Mixing.StartPulay」で指定します。
これらの方法を開始するまでのSCFステップでは単純混合法あるいはKerker混合法が使用されます。
デフォルトでは6に設定されています。
scf.Mixing.EveryPulay
Pulayタイプの混合法における残差ベクトルは、SCFステップが進むに従い、相互に線形従属になる傾向にあり、このため収束が困難になります。
線形従属を避ける方法のひとつは、Kerker混合の合間に時々Pulayタイプの混合を挿入するというものです。
この方法におけるPulayタイプ混合の使用頻度を、「scf.Mixing.EveryPulay」キーワードで指定します。
例えば「scf.Mixing.EveryPulay=5」と設定した場合、SCF反復5回につき1回のPulay混合が実施され、
その他の反復ではKerker法が使用されます。「scf.Mixing.EveryPulay=1」は従来のPulayタイプ混合と同じものになります。
このキーワード
「scf.Mixing.EveryPulay」は、「RMM-DIISK」でのみサポートされており、デフォルト値は1です。
scf.criterion
SCF計算の収束条件を「scf.criterion」キーワードで指定します(Hartree単位)。
SCF反復は、
dUelescf.criterionという条件が満たされると終了します。
ここでdUeleとは、現在のSCF反復とひとつ前の反復とのバンドエネルギーの絶対差です。
デフォルト値は1.0e-6(Hartree単位)です。
scf.Electric.Field
「scf.Electric.Field」キーワードによって、のこぎり波形で表される外部均一電場を導入することが可能です。
例えば、a軸に沿って1.0 GV/m (10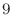 V/m)の電場を適用するには、次のように指定します。
V/m)の電場を適用するには、次のように指定します。
scf.Electric.Field 1.0 0.0 0.0 # default=0.0 0.0 0.0 (GV/m)
電場の符号は電子に作用するものとして定義されています。デフォルト値は「0.0 0.0 0.0」です。
scf.system.charge
電子および正孔のドーピングの量を「scf.system.charge」キーワードで指定します。プラス・マイナス符号はそれぞれ正孔と電子のドーピングを表します。
デフォルト値は0です。
scf.SpinOrbit.Coupling
スピン軌道相互作用を有効にする場合にはこのキーワードを「ON」に設定し、それ以外では「OFF」に設定して下さい。
スピン軌道相互作用を使用する場合、ノンコリニアDFT法の枠組みにおいてj依存擬ポテンシャルを使用しなければなりません。
これについては「相対論的効果」の章も参照して下さい。
1D FFT
1DFFT.EnergyCutoff
局在擬原子軌道と非局所ポテンシャルのプロジェクタの動径関数をフーリエ変換するエネルギー領域を
「1DFFT.EnergyCutoff」キーワードで指定します。このエネルギー領域はフーリエ空間における動径方向
の最大値となります。デフォルト値は3600(Ryd)です。
1DFFT.NumGridK
0から1DFFT.EnergyCutoffまでのエネルギー領域は「1DFFT.NumGridK」キーワードで指定されるグリット数で離散化されます。
局在擬原子軌道および非局所ポテンシャルのプロジェクタの動径関数のフーリエ変換された値は、k空間上の動径方向の
関数として、0から1DFFT.EnergyCutoffまでの範囲で、このグリット上で保持されています。デフォルト値は900です。
1DFFT.NumGridR
実空間上の動径グリット数を「1DFFT.NumGridR」キーワードで指定します。
この値は局在擬原子軌道および非局所ポテンシャルのプロジェクタの動径関数をフーリエ変換する際の数値積分に使用されます。
デフォルト値は900です。
軌道最適化
orbitalOpt.Method
軌道最適化の方法を「orbitalOpt.Method」キーワードで指定します。軌道最適化を行わない場合はこの値を「OFF」に設定して下さい。
軌道最適化を行う場合は、次のオプションから選択します。「atoms」を指定した場合、各原子単位の基底関数が最適化されます。
「species」を指定した場合、各原子種の基底関数が最適化されます。
「atom」および「species」のどちらの場合でも、動径波動関数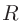 は磁気量子数には依存しないという条件のもとで最適化されます。
これにより最適化基底関数を用いた計算において、全エネルギーの回転不変性が保証されます。
「atom」を指定した場合には最適化された基底関数は化学的環境に依存するため、同一原子種であっても原子毎に異なります。
一方、「species」を指定した場合には「Definition.of.Atomic.Species」で定義した同一名の原子種の基底関数は同一の軌道に最適化されます。
ほぼ同様の化学的環境を持つ原子に対して同一の最適化基底を生成したい場合に、有用な方法です。
は磁気量子数には依存しないという条件のもとで最適化されます。
これにより最適化基底関数を用いた計算において、全エネルギーの回転不変性が保証されます。
「atom」を指定した場合には最適化された基底関数は化学的環境に依存するため、同一原子種であっても原子毎に異なります。
一方、「species」を指定した場合には「Definition.of.Atomic.Species」で定義した同一名の原子種の基底関数は同一の軌道に最適化されます。
ほぼ同様の化学的環境を持つ原子に対して同一の最適化基底を生成したい場合に、有用な方法です。
orbitalOpt.scf.maxIter
軌道最適化におけるSCF反復の最大回数を「orbitalOpt.scf.maxIter」キーワードで指定します。
orbitalOpt.Opt.maxIter
軌道最適化の反復の最大回数を「orbitalOpt.Opt.maxIter」キーワードで指定します。軌道最適化の反復は、収束条件が達成しなかった場合でも、同キーワードで設定した回数で終了します。
orbitalOpt.Opt.Method
軌道最適化の収束方法として、2つの手法がサポートされています。
「EF」は固有ベクトル追跡法、「DIIS」は反復部分空間における直接反転法です。それぞれのアルゴリズムは構造最適化のそれと同じです。「orbitalOpt.Opt.Method」キーワードでは「EF」あるいは「DIIS」を指定して下さい。
orbitalOpt.StartPulay
「orbitalOpt.StartPulay」キーワードで指定した最適化ステップから、準ニュートン法である「EF」または「DIIS」法を開始します。
orbitalOpt.HistoryPulay
準ニュートン法である「EF」および「DIIS」法において、次ステップでの縮約係数を推定するために参照する過去の
ステップ数を「orbitalOpt.HistoryPulay」キーワードで指定します。
orbitalOpt.SD.step
準ニュートン法である「EF」および「DIIS」法を開始するまでの最適化ステップは最急降下法が適用されます。
最急降下法で使用する前因子は「orbitalOpt.SD.step」キーワードで指定します。
多くのケースにおいて、「orbitalOpt.SD.step」の適切な値は0.001程度となります。
orbitalOpt.criterion
軌道最適化の収束条件((Hartree/bohr)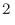 )を「orbitalOpt.criterion」キーワードで指定します。
「微分のノルムorbitalOpt.criterion」という条件が満たされた時に反復ループが終了します。
)を「orbitalOpt.criterion」キーワードで指定します。
「微分のノルムorbitalOpt.criterion」という条件が満たされた時に反復ループが終了します。
CntOrb.fileout
最適化動径関数をファイルに出力したい場合は、「CntOrb.fileout」キーワードを「ON」にする必要があります。
Num.CntOrb.Atoms
最適化動径関数をファイルに出力する際の原子数を「Num.CntOrb.Atoms」キーワードで指定します。
Atoms.Cont.Orbitals
「Atoms.SpeciesAndCoordinates」キーワードの第1列で定義した原子の通し番号を用いて、
最適化基底を出力する原子を「Atoms.Cont.Orbitals」キーワードで次のように指定します。
<Atoms.Cont.Orbitals
1
2
Atoms.Cont.Orbitals>
記述は「Atoms.Cont.Orbitals」で始め、「Atoms.Cont.Orbitals」で終わります。
行の数は、
「Atoms.Cont.Orbitals」で記述する数字と整合性がなければなりません。
例えば、最適化擬原子軌道が「C_1.pao」と「H_2.pao」などとして保存された場合、その原子種名はキーワード
「Definition.of.Atomic.Species」の
設定における第一列の記号に対応し、記号の後の数字はキーワード「Atoms.SpeciesAndCoordinates」の設定における第一列の数値を意味します。
これらの出力ファイル「C_1.pao」と「H_2.pao」は基底関数の入力データとして使用可能です。
O() SCF計算
orderN.HoppingRanges
各原子を中心とする球の半径(Å)を「orderN.HoppingRanges」キーワードで定義します。
DC法、DC-LNO法およびO()クリロフ部分空間法では、この球内に含まれる原子を選択することで切り取られたクラスタが構成されます。
orderN.KrylovH.order
切り取られた各クラスタのハミルトニアンに対し、クリロフ部分空間の次元を「orderN.KrylovH.order」キーワードで指定します。
orderN.KrylovS.order
後述のキーワードで「orderN.Exact.Inverse.S=off」に設定した場合、重なり積分の逆行列は
クリロフ部分空間法を用いて近似されます。
この時、切り取られた各クラスタに対する重なり行列のクリロフ部分空間法の次元を「orderN.KrylovS.order」キーワードで指定します。
デフォルト値は、「orderN.KrylovH.order」で設定した値の4倍です。
orderN.Exact.Inverse.S
キーワード「orderN.Exact.Inverse.S」を「on」に設定すると、切り取られた各クラスタの重なり行列の逆行列は厳密に評価されます。
offに設定する場合は前述のキーワード「orderN.KrylovS.order」の項を参照して下さい。
デフォルトでは「on」に設定されています。
orderN.Recalc.Buffer
キーワード「orderN.Recalc.Buffer」を「on」に設定すると、バッファ行列は各SCF反復毎に再計算されます。
「off」の場合にはバッファ行列は初回のSCFステップで計算され、その後のSCF反復では固定されます。
デフォルトでは「on」に設定されています。
orderN.Expand.Core
キーワード「orderN.Expand.Core」を「on」に設定すると、コア領域は半径
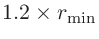 の球の中にある
原子から構成されます。ここで
の球の中にある
原子から構成されます。ここで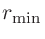 は、中心原子と最隣接原子間の距離です。
このコア領域はクリロフ部分空間を生成するときの第1ステップで使用されるベクトル群を定義します。
「orderN.Expand.Core」が「off」の場合、中心の原子がコア領域と見なされます。デフォルトでは「on」に設定されています。
は、中心原子と最隣接原子間の距離です。
このコア領域はクリロフ部分空間を生成するときの第1ステップで使用されるベクトル群を定義します。
「orderN.Expand.Core」が「off」の場合、中心の原子がコア領域と見なされます。デフォルトでは「on」に設定されています。
MD(分子動力学)あるいは構造最適化
MD.Type
分子動力学計算あるいは構造最適化のタイプを指定します。
現在使用できるオプションは以下の通りです。
MD無し(「Nomd」)、NVEアンサンブルMD(「NVE」)、速度スケーリング法によるNVTアンサンブルMD(「NVT_VS」)[30]、Nose-Hoover法によるNVTアンサンブルMD(「NVT_NH」)[31]、複数熱浴付きMD(「NVT_VS2」あるいは「NVT_VS4」)、最急降下法による構造最適化(「SD」)、DIIS最適化法(「DIIS」)、固有値ベクトル追跡法(「EF」)[63]、及び合理的関数法(「RF」)[64]です。
その他にセル最適化のためのオプション(OptC1、OptC2、OptC3、OptC4、OptC5、OptC6、OptC7、RFC5、RFC6、RFC7)も
利用可能です。
詳細は「構造最適化」、「セル可変構造最適化」および「分子動力学」の章を参照して下さい。
MD.Fixed.XYZ
次のキーワードを指定することで、構造最適化および分子動力学シミュレーションの際に任意の原子座標を初期座標に固定することができます。
各原子の、、座標に対して、以下のように設定します。
<MD.Fixed.XYZ
1 1 1 1
2 1 0 0
MD.Fixed.XYZ>
上記の例は2つの原子から構成される系の場合です。系に個の原子がある場合、個分の指定が必要です。
第1列は「Atoms.SpeciesAndCoordinates」で個々の原子を特定するのに使用した連番です。
第2〜4列は、それぞれ、、座標のフラグです。フラグが「1」だと座標は固定、「0」だと緩和されます。
上記の例では、原子1の、、座標は固定、原子2は座標のみ固定されています。デフォルトでは全ての座標が緩和される指定になっています。
このキーワードによる原子座標の固定は、すべての構造最適化および分子動力学の方法において有効です。
MD.maxIter
MD計算や構造最適化計算における最大の反復回数を「MD.maxIter」キーワードで指定します。
MD.TimeStep
時間ステップ(fs)を「MD.TimeStep」キーワードで指定します。
MD.Opt.criterion
「MD.Type」キーワードによって構造最適化の方法を選択している場合には、「MD.Opt.criterion」キーワードでその収束条件(Hartree/Bohr)
を設定します。原子にかかる力の最大絶対値が「MD.Opt.criterion」で指定した値より小さくなった場合に、構造最適化は終了します。
MD.Opt.DIIS.History
「DIIS」、「EF」、「RF」による構造最適化を行う場合、「MD.Opt.DIIS.History」キーワードは構造最適化のために
参照する過去のステップ数を指定します。デフォルト値は3です。
MD.Opt.StartDIIS
「DIIS」、「EF」、「RF」による構造最適化を開始するステップを「MD.Opt.StartDIIS」キーワードで指定します。
DIISタイプの構造最適化を開始する以前のステップでは最急降下法が使用されます。デフォルト値は5です。
MD.TempControl
キーワード「MD.TempControl」によりNVTアンサンブルの分子動力学計算における原子運動の温度を指定します。
「NVT_VS」を選択した場合、原子運動の温度を下記の例のように制御できます。
<MD.TempControl
3
100 2 1000.0 0.0
400 10 700.0 0.4
700 40 500.0 0.7
MD.TempControl>
記述は「MD.TempControl」で開始し、「MD.TempControl」で終わります。最初の数字「3」は、続く温度指定の行数を指定します。
例では3行あります。後続する行の第1列はMDステップ数を意味し、第2列は速度スケーリングを行うMDステップの間隔を指定します。
例では、100ステップ目までは2ステップ毎に、100〜400ステップ間は10ステップ毎に、400〜700ステップ間は40ステップ毎に速度スケーリングを行います。
第3、4列はそれぞれ温度(K)とスケーリングパラメータ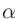 を指定します。
詳細は「分子動力学」の章を参照して下さい。
一方、NVT_NHの場合、以下の記述で原子運動の温度を制御できます。
を指定します。
詳細は「分子動力学」の章を参照して下さい。
一方、NVT_NHの場合、以下の記述で原子運動の温度を制御できます。
<MD.TempControl
4
1 1000.0
100 1000.0
400 700.0
700 600.0
MD.TempControl>
記述は「MD.TempControl」で開始し、「MD.TempControl」で終わります。
最初の数字「4」は、続く温度指定の行数を指定します。
この例では4行あります。
後続する行の第1、2列は、それぞれMDステップ数と原子運動の温度を指定します。
指定されたMDステップ間の温度は線形補完されます。
NH.Mass.HeatBath
キーワード「MD.Type」によって「NVT_NH」を選択した場合、このキーワードで熱浴の質量を設定します。
デフォルト値は20です。次元は長さ 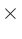 質量です。
この表記では長さにはボーア半径、質量に統一原子質量単位(炭素原子の主同位体の質量を12.0とする単位)を用います。
質量です。
この表記では長さにはボーア半径、質量に統一原子質量単位(炭素原子の主同位体の質量を12.0とする単位)を用います。
MD.Init.Velocity
分子動力学のシミュレーションでは、下記の記述により各原子に初期速度を与えることができます。
<MD.Init.Velocity
1 3000.000 0.0 0.0
2 -3000.000 0.0 0.0
MD.Init.Velocity>
これは2つの原子からなる系の例です。個の原子がある場合、同様に行の記述が必要です。
第1列は「Atoms.SpeciesAndCoordinates」キーワードで指定した原子の連番と同じです。第2、3、4列は、各原子の速度の、、成分で、単位はm/sです。
「MD.Init.Velocity」キーワードは、「MD.Fixed.XYZ」キーワードと併用できます。
バンド分散
Band.dispersion
バンド分散を評価する場合は「Band.dispersion」キーワードを「ON」に設定して下さい。
Band.KPath.UnitCell
「Band.KPath.UnitCell」キーワードによって、下記の例のようにバンド分散の計算で使用する単位胞ベクトルを指定します。
<Band.KPath.UnitCell
3.56 0.0 0.0
0.0 3.56 0.0
0.0 0.0 3.56
Band.KPath.UnitCell>
記述は「Band.KPath.UnitCell」で開始し、「Band.KPath.UnitCell」で終了します。
「Band.KPath.UnitCell」キーワードの記述がある場合、同キーワードで指定した単位胞ベクトルからバンド分散の
計算に使用する逆格子ベクトルを算出します。
「Band.KPath.UnitCell」を指定しない場合には、「Atoms.UnitVectors」キーワードで指定した単位胞ベクトルから
計算した逆格子ベクトルが使用されます。面心立方格子構造 (fcc)や体心立方格子構造(bcc)の場合には
「Band.KPath.UnitCell」キーワードで逆格子ベクトルを再定義した方が、特殊k点の指定が容易です。
Band.Nkpath
バンド分散の計算における経路の数を「Band.Nkpath」で指定します。
Band.kpath
バンド分散の経路を「Band.kpath」キーワードで下記のように指定します。
<Band.kpath
15 0.0 0.0 0.0 1.0 0.0 0.0 g X
15 1.0 0.0 0.0 1.0 0.5 0.0 X W
15 1.0 0.5 0.0 0.5 0.5 0.5 W L
15 0.5 0.5 0.5 0.0 0.0 0.0 L g
15 0.0 0.0 0.0 1.0 0.0 0.0 g X
Band.kpath>
記述は「Band.kpath」で始まり、「Band.kpath」で終わります。
行数は「Band.Nkpath」で指定した数と同じでなくてはなりません。
第1列には経路上で固有値を計算する格子の数を指定します。
続く(n1, n2, n3)および(n1', n2', n3')は、逆格子ベクトルを基底とする第1ブリルアンゾーンの経路の開始および
終了のk点を指定します。
「Band.KPath.UnitCell」キーワードを指定している場合、バンド分散を計算するための逆格子ベクトルは
「Band.KPath.UnitCell」で設定した単位胞ベクトルを用いて計算されます。
「Band.KPath.UnitCell」の定義が無い場合は「Atoms.UnitVectors」で指定した単位胞ベクトルを用いて逆格子ベクトルを計算します。
最後の2つの英文字は経路の開始および終了のk点の名前を指定します。
再スタート
scf.restart
過去の計算で生成されたリスタートファイル「System.Name_rst/*」を用いてSCF計算を再スタートさせる場合には、
このキーワード「scf.restart」を「ON」に設定して下さい。
分子軌道(MO)の出力
MO.fileout
分子軌道をファイルに出力したい場合は、キーワード「MO.fileout」を「ON」に設定して下さい。
num.HOMOs
出力する最高被占分子軌道(HOMO)の数を「num.HOMOs」キーワードで指定します。
num.LUMOs
出力する最低空分子軌道(LUMOS)の数を「num.LUMOs」キーワードで指定します。
MO.Nkpoint
「MO.fileout=ON」および「scf.EigenvalueSolver=Band」を設定している場合、「MO.Nkpoint」キーワードでMOを出力するk点の数を指定します。
MO.kpoint
MOを出力する k点を「MO.kpoint」キーワードで以下のように指定します。
<MO.kpoint
0.0 0.0 0.0
MO.kpoint>
記述は「MO.kpoint」で開始し、「MO.kpoint」で終わります。k点は逆格子ベクトルを基底して(n1, n2, n3)の3つの数字で指定します。
ここで、逆格子ベクトルは「Band.kpath」の場合と同様な方法で生成されたものが使用されます。
DOSとPDOS
Dos.fileout
全状態密度(DOS)および射影した部分状態密度(PDOS)を評価する場合は「Dos.fileout」キーワードを「ON」に設定して下さい。
Dos.Erange
DOS計算におけるエネルギー範囲を「Dos.Erange」キーワードで下記のように指定します。
Dos.Erange -10.0 10.0
ここで2つの値はそれぞれエネルギー範囲の下限と上限(eV)です。
Dos.Kgrid
DOS計算を行う上で、第1ブルリアンゾーンを離散化するために(n1, n2, n3)の格子点を「Dos.Kgrid」キーワードで指定します。
開発者向けインタフェース
HS.fileout
後処理計算において、Kohn-Shamハミルトニアン行列、重なり行列、密度行列を利用する場合には「HS.fileout」キーワードを「ON」に設定して下さい。
これによりバイナリ形式のデータが「System.Name.scfout」に格納されます。ここで、「System.Name」はキーワード「System.Name」で設定したファイル名です。
このデータの利用に関しては「開発者向けインタフェース」の章で詳しく説明します。
Voronoi電荷
Voronoi.charge
Voronoi電荷を計算する場合には「Voronoi.charge」キーワードを「ON」に設定して下さい。
計算結果は「System.Name.out」ファイルに保存されます。ここで「System.Name」は「System.Name」で設定したファイル名です。
Next: 出力ファイル
Up: 入力ファイル
Previous: 例:メタン分子
Contents
Index