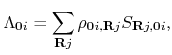Next: Krylov subspace method Up: Order(N) method Previous: Divide-conquer method Contents Index
The DC-LNO method [51] is a variant of the DC method. The computational efficiency is improved by introducing localized natural orbitals (LNOs) to span the subspace of each atom. The dimension of the resultant subspace is smaller than that in the DC method, leading to the reduction of computational cost. The LNOs are non-iteratively calculated by a low-rank approximation via a local eigendecomposition of a projection operator for the occupied space. As shown in Fig. 19(a), introducing LNOs to represent the long range region of a truncated cluster reduces the computational cost of the DC method while keeping computational accuracy. The method can be applied to not only gapped systems, but also metallic systems as long as the size of truncated clusters is large enough, and typically clusters including more than 200 atoms might be chosen. The functionality is compatible with not only the collinear calculation, but also the non-collinear calculations.
![\includegraphics[width=16.0cm]{DC-LNO-Fig1.eps}](img169.png)
|
As a first step for O(![]() ) calculations of the DC-LNO method, one can perform
an O(
) calculations of the DC-LNO method, one can perform
an O(![]() ) calculation for Si crystal using an input file store in the directory 'work' as
) calculation for Si crystal using an input file store in the directory 'work' as
% mpirun -np 112 ./openmx Si8-LNO.dat | tee si8-lno.std
The calculation was performed in 66 seconds using 112 cores on a Xeon cluster machine of 2.6 GHz.
In order for you to start an trial calculation by the DC-LNO method, an input file 'Si8-LNO.dat' is
available in the directory 'work'.
Since as shown in Fig. 19(b) the three level parallelization has been implemented:
atom level, spin level, and diagonalization level, it is expected that the method scales up to, e.g.,
40000 CPU cores for a 1000 atom system
in the parallel calculations, where we assumed 1000 atoms
scf.dclno.threading on # off|on
In the hybrid parallelization the diagonalization at the bottom level will be parallelized by OpenMP.
The computational accuracy and efficiency of the method can be controlled by the following keywords:
orderN.HoppingRanges 7.0 # 7.0 (Ang.)
orderN.LNO.Buffer 0.2 # default = 0.2
orderN.LNO.Occ.Cutoff 0.1 # default = 0.1
The role of the keyword 'orderN.HoppingRanges' is exactly the same as that in the DC method.
For each atom a truncated cluster is constructed by picking-up atoms within a sphere whose radius is
specified by the keyword 'orderN.HoppingRanges'. Though the proper choice of the parameter depends on
systems, a serie of benchmark calculations implies that the accuracy is enough for not only gapped systems,
but also metallic systems if 'orderN.HoppingRanges' is set so that the resultant truncated cluste can
include 300 atoms. The setting might be regarded as a conservative choice to ensure the accuracy rather
than efficiency. So, a compromising choice with respect to both accuracy and efficiency may be
in between 200 and 300 atoms. The region in the truncated cluster where the PAOs are replaced by LNOs
is determined by the keyword 'orderN.LNO.Buffer'. The 'orderN.LNO.Buffer=0.0' means that PAOs allocated
on all the SNAN atoms are replaced by LNOs, while the PAOs on all the SNAN atoms remain unchanged
in the case with the 'orderN.LNO.Buffer=1.0' which is equivalent to the DC method.
As for the SNAN, please refer the subsection 23.4 'User definition of FNAN+SNAN'.
![\includegraphics[width=15.9cm]{DC-LNO-Fig2.eps}](img170.png)
|
<Definition.of.Atomic.Species
Si Si7.0-s2p2d1 Si_PBE19
H H6.0-s2p1 H_PBE19
Definition.of.Atomic.Species>
the number of LNOs can be specified by
<LNOs.Num
Si 4
H 1
LNOs.Num>
In this case, the numbers of the LNOs are fixed to 4 and 1 for Si and H, respectively.
To avoid a sudden change of the number of LNOs during geometry optimization and molecular dynamics simulations,
it might be better to use 'LNOs.Num' rather than orderN.LNO.Occ.Cutoff. The comparison between the DC and DC-LNO methods
is shown in Fig. 20. Although the PAOs in the long range region are replaced by the LNOs, it is found
that the accuracy is comparable to the DC method both in gapped and metallic systems.
As an illustration for applications of the DC-LNO method, we show in Fig. 21 radial distribution functions (RDF)
of liquids for silicon, aluminum, lithium, and SiO
![\includegraphics[width=14.0cm]{DC-LNO-Fig3.eps}](img177.png)
|
In Fig. 22 the speed-up ratio in the MPI parallelization of the DC-LNO method is shown for non-spin
polarized calculations of a diamond supercell containing 64 atoms. Since the multiplicity of spin index is 1,
we see a nearly ideal behavior up to 64 MPI processes. Beyond 64 MPI processes the parallelization in the diagonalization
level is taken into account on top of the parallelization in the atom level.
![\includegraphics[width=11.0cm]{DC-LNO-Fig4.eps}](img178.png)
|