The DFT-D3 method of Grimme et al. [92,93] is supported to include a vdW
interaction with default parameters for the GGA-PBE functional. The following keywords
are relevant for the DFT-D3 method.
scf.dftD on # on|off, default=off
version.dftD 3 # 2|3, default=2
DFTD3.damp bj # zero|bj, default=bj
DFTD.Unit AU # Ang|AU
DFTD.rcut_dftD 100.0 # default=100 (DFTD.Unit)
DFTD.cncut_dftD 40 # default=40 (DFTD.Unit)
DFTD.IntDirection 1 1 1 # default=1 1 1 (1:on 0:off)
When you include the DFT-D2 or DFT-D3 calculation, turn on 'scf.dftD'.
For DFT-D2 use version.dftD=2 and for DFT-D3 version.dftD=3. The DFT-D3
implemented here supports both zero and Becke-Johnson (BJ) damping
functions [93]. The cutoff radius for the interaction is given
by 'DFTD.rcut_dftD' and for the coordination number calculation 'DFTD.cncut_dftD'.
The units are given by 'DFTD.Unit' and the suggested defaults for
both cutoff values are in AU. Also, the interaction for image atoms
can be cut along the a-, b-, and c-axes by 'DFTD.IntDirection', where
1 means that the interaction is included, and 0 not. Also, the periodicity
for each atom can be controlled as in the case of the DFT-D2 method by
<DFTD.periodicity
1 1
2 1
3 1
4 1
....
DFTD.periodicity>
where the first column is a serial number which is the same as in
the 'Atoms.SpeciesAndCoordinates', and the second column is a flag
which means that 1 is periodic, and 0 is non-periodic for the corresponding
atom. By considering the periodicity or non-periodicity of each atom,
the interaction is automatically cut when they are non-periodic.
The main modifications are placed at only two routines: DFTD3vdW_init.c
and Calc_EdftD() of Total_Energy.c. In DFTD3vdW_init.c, you can
easily change the parameters for the vdW correction, and in Calc_EdftD3()
of Total_Energy.c you can confirm how they are calculated.
Parameters for other functionals may be set through the following keywords
DFTD.scale6 1 # default=0.75|1.0 (for DFT-D2|DFT-D3)
DFTD.scale8 0.7875 # default=0.722|0.7875 (for PBE with zero|bj damping)
DFTD.sr6 1.217 # default=1.217 (for PBE)
DFTD.a1 0.4289 # default=0.4289 (for PBE)
DFTD.a2 4.4407 # default=4.4407 (for PBE)
The '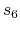 ' and '
' and '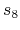 ' global scaling value of Eq. (3) in Grimme's
paper [92] is given by 'DFTD.scale6' and 'DFTD.scale8'.
The global scaling parameters are functional and damping-function dependent.
The parameter 'sr6' of Eq. (6) in [92] needs to be set when using
the zero damping function while the parameters 'a1' and 'a2' of Eq.
(6) in [93] need to be set when choosing BJ damping.
' global scaling value of Eq. (3) in Grimme's
paper [92] is given by 'DFTD.scale6' and 'DFTD.scale8'.
The global scaling parameters are functional and damping-function dependent.
The parameter 'sr6' of Eq. (6) in [92] needs to be set when using
the zero damping function while the parameters 'a1' and 'a2' of Eq.
(6) in [93] need to be set when choosing BJ damping.
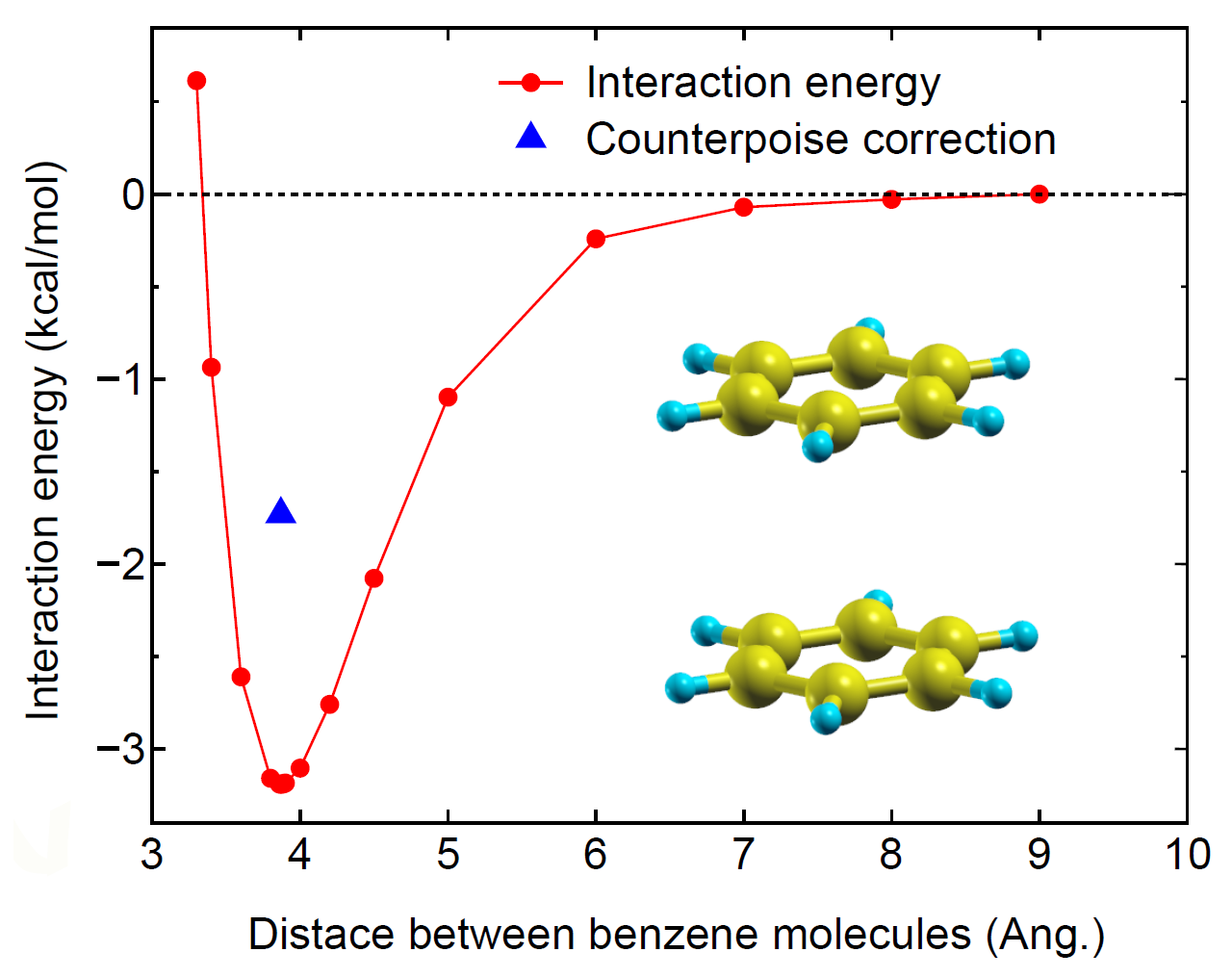
As an example for the DFT-D3 calculation, the interaction energy between
two benzene molecules in a parallel structure with 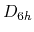 symmetry is shown
as a function of the inter-distance in Fig. 49.
All the input files for the calculations can be found in a directory 'work/DFT-D3/',
and they are
symmetry is shown
as a function of the inter-distance in Fig. 49.
All the input files for the calculations can be found in a directory 'work/DFT-D3/',
and they are
Dimer-Ben-10.0.dat Dimer-Ben-3.88.dat Dimer-Ben-4.5.dat Mono-Ben-1.dat
Dimer-Ben-3.3.dat Dimer-Ben-3.89.dat Dimer-Ben-5.0.dat Mono-Ben-2.dat
Dimer-Ben-3.4.dat Dimer-Ben-3.8.dat Dimer-Ben-6.0.dat Mono-Ben.dat
Dimer-Ben-3.6.dat Dimer-Ben-3.9.dat Dimer-Ben-7.0.dat
Dimer-Ben-3.86.dat Dimer-Ben-4.0.dat Dimer-Ben-8.0.dat
Dimer-Ben-3.87.dat Dimer-Ben-4.2.dat Dimer-Ben-9.0.dat
After optimizing the monomer using 'Mono-Ben.dat', the total energy of dimer in
a variety of inter-distance was calculated using 'Dimer-Ben-#.dat' (#=3.3-9.0),
where the structure of the benzene molecule is the same as the structure of monomer
obtained by the first calculation. The monomer calculations with a counterpoise
correction were performed by 'Mono-Ben-1.dat' and 'Mono-Ben-2.dat'.
The optimum inter-distance is found to be 3.87 Å, which is well compared with
a reported value (3.89 Å) computed with density fitted local second-order Møller–Plesset
perturbation theory (DF-LMP2) [94]. The counterpoise corrected interaction energy
is 1.73 kcal/mol being in good agreement with a reported value (1.7 kcal/mol) [94],
while the basis set superposition error is found to be large.
Figure 49:
The interaction energy of between two benzene molecules in a parallel structure with symmetry.
The counterpoise corrected interaction energy is shown by the triangle.
All the input files for the calculations can be found in a directory 'work/DFT-D3/.
 |
2016-04-03