The primitive and optimized PAO functions are usually assigned to atoms. Moreover, it is possible to assign basis functions in any vacant region using an empty atom. You will find the empty atom 'E' in the web site of the database (http://www.openmx-square.org/). Using the pseudopotential for the empty atom 'E', though the pseudopotential is a flat zero potential, you can put basis functions at any place independently of atomic position. To do that, you can define empty atoms by
<Definition.of.Atomic.Species
H H5.0-s2p1 H_PBE13
C C5.0-s2p1 C_PBE13
EH H5.0-s2p1 E
EC C5.0-s2p1 E
Definition.of.Atomic.Species>
In the example, two sorts of empty atoms are defined as 'EH' and 'EC' which have
basis sets specified by 'H5.0-s2p1' and 'C5.0-s2p1', respectively, which means that
one can use any basis functions for an empty atom as shown above. Then 'EH' and 'EC'
can be put to any place by the keyword 'Atoms.SpeciesAndCoordinates', where the number
of electrons for the empty atom is zero. To define an empty atom, only thing you have
to do is to use 'E.vps' as pseudopotential for the empty atom.
The empty atom scheme enables us not only to estimate the basis set superposition error (BSSE)
using the counterpoise correction (CP) method [33,34], but also to treat
a vacancy state and a nearly free electron state on metal surfaces within
the linear combination of pseudo-atomic orbitals (LCPAO) method.
As an example, a calculation of a F-center in NaCl with a Cl vacancy is shown in Fig. 3.
We see that the highest occupied state at the
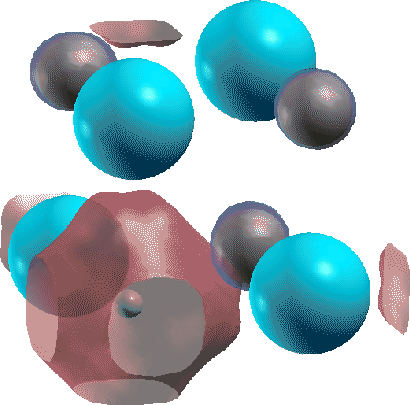 |